
Parmi ces espèces certaines se trouvent en général en concentrations faibles. Leur dosage ne présente donc pas un intérêt marqué pour ce devoir. Nous nous intéresserons aux espèces du soufre présentent dans un domaine de pH compris entre 5 et 10, ces de la plus part des eaux sulfurées. Le soufre élémentaire s'il est en état dissous sera en concentration très faible et n'interviendra pas dans le bilan des espèces dissoutes. Si l'on est en présence de soufre colloïdal, une extraction par solvant ( trichloroéthylène, chloroforme, ...) ou une filtration sur filtre de pores de tailles égales à 0.01µm permettra de le séparer des espèces dissoutes et de le doser à part. Nous n'examinerons pas non plus le dosages des sulfates.
Dans ce qui suit nous allons examiner les méthodes traditionnelles de dosages, puis des méthodes basées sur l'emplois de l'électrode spécifique des sulfures et enfin comparer les résultats acquis sur les eaux naturelles et en particulier sur les eaux sulfurées.
I Dosage des sulfures
A l'émergence de la source on mesure le pH et la réserve alcaline ( après addition de HCl).Ces mesures sont particulièrement importantes pour l'exploitation ultérieure des analyses. Les eaux sulfurées des Pyrénées sont en général situées à des pH entre 8.7 et 10; à ces pH la silice est partiellement ionisée selon la réaction
H4SiO4 --® H+ + H3SiO4-
et donc elle contribue à la réserve alcaline mesurée. C'est aussi le cas des sulfures qui sont sous la forme HS-. La réserve alcaline (RB) qui correspond à la somme de la concentration des cations et des anions (exprimée en équivalent/litre) n'ayant pas de propriétés acido-basiques.
En tenant compte de la relation d'électro-neutralité on peut donc écrire dans le cas des eaux pyrénéennes:
RB = [HCO-] + 2 [CO32-] + [H3SiO4-] + [SH-] + [OH-] où [i] désigne la
concentration de l'espèce [i]. L'estimation de la contribution de H3SiO4- se fait à partir de la mesure de la concentration en silice dissoute et du pH. De façon analogue on déduit SH- de la concentration totale en hydrogène sulfuré et du pH. Afin d'éviter une accumulation d'incertitude dans l'estimation des carbonates, la concentration en carbonate dissous est mesurée par chromatographie en phase gazeuse, ce qui permet de vérifier la cohérence des analyses chimiques avec la réserve alcaline mesurée.
Les espèces du soufre autres que les sulfates sont déterminées par dosage au chlorure mercurique avec une électrode spécifique des ions S2-[2,3,4].Ces dosages sont effectués sous azote afin d'éviter toute oxydation après le prélèvement.
La silice, les sulfates et l'ammoniac sont dosés dans les heures qui suivent le prélèvement. Les sulfates sont dosés par titration avec BaCl2 au moyen d'une électrode spéciale des ions Ba+ , ou par conductimétrie.
Des échantillons sont filtrés (0.1 et 0.01 m ) et acidifiés à pH=2. Sur ces échantillons les analyses des cations et anions majeurs, ainsi que des éléments traces sont faites au laboratoire dans les jours qui suivent le prélèvement. Les méthodes employées sont principalement l'absorption atomique en flamme (Na, K, Mg, Ca) ou sans flamme (Li,Rb,Cs,Sr,Ba,Mn,Fe,Cu) ou des méthodes électrochimiques (F, B).
a) Méthodes de dosage basés sur l'oxydation des espèces du soufre par l'iode
Ces méthodes sont classiques et figurent dans de nombreux ouvrages de chimie analytique. En milieu acide ( tampon acétate) on a les réactions suivantes entre l'iode et les espèces du soufre:
H2S + I2 --® I- + H+
Sn2- + I2 --® n S + 2 I-
2 S2O32- + I2 --® S4+O62- + 2 I-
HSO3- + I2 + H2O --® SO42- + 3 H+ + 2 I-
La réaction de l'iode avec une eau sulfurée permettra de connaître
S [H2S] +S [Sn2-] + 1/2 [S2O32-] + S [ SO32-]
En complexant le sulfite par le formaldéhyde, on bloque sa réaction avec l'iode; donc un dosage en présence de formaldéhyde donnera S [H2S] +S [Sn2-] + 1/2 [S2O32-].Le soufre sulfure peut être précipité à l'état de sulfure métallique par addition de sels de zinc ou de cadmium; le précipité est séparé par filtration sous azote. Ensuite on peut doser 1/2 [S2O32-] + S [ SO32-] par l'iode, et sur un autre aliquot [S2O32-] après addition de formaldéhyde. Le précipité de sulfure de zinc ou de cadmium est ensuite redissout par additionne HCl et on dose par l'iode; d'où on en déduit S [H2S] +S [Sn2-]. Dans la pratique on ajoute la solution où l'eau à doser en excès d'iode que l'on dose par le thiosulfate en suivant la réaction potentiométriquement.
L'ensemble des résultats de ces dosages doit être cohérent. La précision de cette méthode est de l'ordre de 1% pour des concentrations supérieures à 10-4 mole.kg-1, et de l'ordre de 2% à 5% pour des concentrations supérieure à 5.10-5 mole.kg-1;elle est beaucoup plus faible pour des concentrations inférieures. La précision est elle aussi plus faible dans les eaux chaudes car les risques d'évaporation ne sont pas négligeables.
Le soufre des polysulfures, ((n-1)[Sn2-], peut être connu par réaction avec les sulfites à chaud selon la réaction:
H+ + Sn2- + (n-1)SO32- --® HS- + (n-1)S2O32- (1)
A la fin de cette réaction on suit les mêmes processus que ci-dessus, d'où l'on déduit le soufre des polysulfures par la mesure de l'excès de thiosulfate formé. Le soufre colloïdal réagit selon:
S + SO32- --® S2O32-
et il doit donc être extrait ou filtré avant de faire cette réaction. Cette méthode de dosage du soufre des polysulfures peut aussi être suivie directement par potentiométrie. On mesure alors simultanément le pH et le potentiel d'un électrode de platine (Eh) et on trace le graphe Eh-pH; la fin du dosage correspond à un saut de potentiel dans ce diagramme. Le soufre total dissout est dosé par la même méthode que le sulfate après oxydation des espèces par addition de soude et d'eau oxygénée à chaud.
Pour obtenir des résultats satisfaisants chaque dosage doit être doublé. Cette méthode par dosages successifs à l'iode est assez longue pour des concentrations individuelles en espèces dosées inférieures à 5.10-4mole.kg-1. De plus si on veut employer ces méthodes avec des eaux sulfurées chaudes comme on en trouve dans les Pyrénées-Orientales par exemple) plusieurs sources d'erreurs vont alors apparaître ou devenir critiques. Tout d'abord du fait de la température les pertes d'iode lors du prélèvement ou par évaporation peuvent ne pas être négligeables. Cette perte d'iode va entraîner une surestimation de la concentration en thiosulfate et en sulfite puisque les sulfures sont estimés indépendamment. De plus dans les eaux chaudes assez alcalines, telles que celles des Pyrénées, la solution d'iode bien que tamponnée vers pH 4-5, est en partie transformée en iodate par hydrolyse, créant ainsi une consommation supplémentaire apparente d'iode, et donc par conséquent une surestimation en thiosulfate et en sulfite. Globalement donc plusieurs causes d'erreurs peuvent s'accumuler et conduire à de nettes surestimations des concentrations en thiosulfate et en sulfite. Ces inconvénients sont majeurs. C'est ce qui a conduit à s'intéresser à de nouvelles méthodes de dosage du soufre en solution.
b) Méthode de dosage basée sur la réaction des espèces du soufre avec l'ion mercurique.
En présence de sels mercurique on observe les réactions suivantes:
en milieu basique (pH=13)
HS- + Hg2- --® HgH + H+ (2)
Sn2- + Hg2+ --® HgS + (n-1) S (3)
2 R S + Hg2+ --® Hg(RS)2 (4)
avec R radical organique
en milieu neutre (pH=7-8)
2 S2O32- + Hg2+ --® Hg(S2O3)22- (5)
2 SO32- + Hg2+ --® Hg(SO3)22- (6)
Ces réactions peuvent donner lieu à une méthode de dosage des espèces réduites
du soufre plus simple et plus rapide que la précédente. A cette fin, il faut disposer d'un moyen de détecter soit les espèces du soufre, soit la consommation d'ions mercuriques. Comme nous allons le voir les électrodes spécifiques des sulfures permettent de résoudre ce problème.
Les électrodes spécifiques des sulfures:
Les électrodes constituées par le couple argent/sulfure d'argent (Ag/Ag2S) sont sensibles et réversibles vis-à-vis des ions S2-. En particulier les électrodes constituées par une membrane en sulfure d'argent sont les plus sensibles et les plus fidèles. Le potentiel obtenu en présence d'hydrogène sulfuré est une fonction de la concentration en ions s2- , soit:
ES2-=E0S2--(R.T.F-1/2)log[S2-].
L'électrode à membrane Ag2S présente aussi une interférence très forte due aux ions mercurite. Cette interférence est aussi réversible. D'où l'idée d'employer l'électrode Ag/Ag2S comme indicatrice des réactions entre espèces du soufre et ions mercurites. En effet les réactions (2) et (3) correspondent à un saut de potentiel dû à la consommation des espèces sulfurées, dans ce cas l'électrode Ag/Ag2S est sensible aux ions S2-. Les mercaptans et autres molécules organiques contenant des atomes de soufre labiles et de degré d'oxydation -2 donnent aussi lieu à un saut de potentiel avec l'électrode Ag/Ag2S. Enfin les réactions (5) et (6) donnent aussi lieu à un saut de potentiel, l'électrode Ag/Ag2S étant alors sensible aux ions Hg2+. A la surface de l'électrode Ag/Ag2S les potentiels respectifs des réactions (2) et (3) sont séparés d'environ 200mV de la réaction (4) et de 500mV à 600mV des réactions (5) et (6), ce qui permet de doser des espèces correspondantes sans séparation préalable.
Processus expérimental:
Les échantillons des solutions ou d'eaux sont introduits dans une cellule électrochimique close et muni de rodages pour les électrodes de pH, Ag/Ag2S, ou de référence ( Ag/AgCl avec un pont électrolytique interne). Des rodages permettent aussi de disposer d'une burette à piston de précision contenant une solution HgCl2 et d'une circulation d'azote. Les échantillons sont prélevés et introduits sous atmosphère d'azote dans la cellule. La cellule est thermostatée et on règle la température à celle de la source étudiée.
J Dosage du soufre et des sulfures ( S[H2S] + S[Sn2-]) et de la somme thiosulfate plus sulfite.
Le pH de l'échantillon est amené à 13 par addition de soude préalablement dégazée avec de l'azote. L'addition d'une solution titrée de HgCl2 entraîne la précipitation de HgS. Un premier saut de potentiel correspondant aux réactions (2) et (3) est obtenu, on en déduit le soufre de sulfure, S[H2S] + S[Sn2-]. Un deuxième saut de potentiel est obtenu si on est en présence de molécules organiques soufrées ( réaction (4)), on en déduit [RS-]. Le pH de la solution est alors ramené entre 7 et 8. Le dosage de la somme thiosulfate plus sulfite est alors effectué selon les réactions (5) et (6).
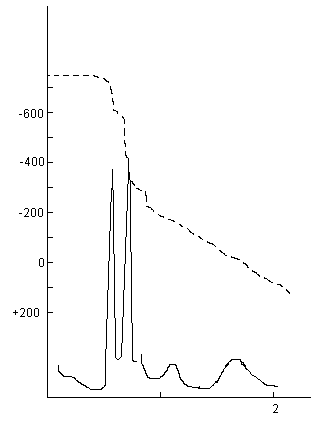
On remarque la présence de deux sauts de potentiels distincts à pH 7: le premier correspond à [S2O32-] et le deuxième à [SO32-]. Pour des concentrations en sulfite inférieure à 10-4 mole.kg-1 ces deux sauts de potentiel sont trop proches pour être exploitables directement. Une deuxième manipulation doit alors être faite après ajout de formaldéhyde.
K Dosage du soufre de sulfure et du thiosulfate.
Le dosage est effectué selon le même processus que le précédent, mais on s'affranchit de la réaction (6) en complexant le sulfite par ajout de formaldéhyde. Du dernier saut de potentiel on déduit [S2O32-] et par différence avec le premier dosage on en déduit [SO32-]. Le premier saut de potentiel doit fournir la même concentration en soufre de sulfure que celle obtenue au premier dosage.
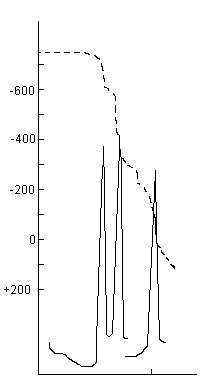
L Dosage du soufre de sulfure et de la somme thiosulfate plus soufre de polysulfure (S(n-1)[Sn2-]).
Un large excès de sulfite (par rapport à la réaction (1)) est ajouté à l'échantillon, puis on laisse réagir les polysulfures avec les sulfites pour donner une quantité équivalente d'hydrogène sulfuré et (ñ-1) fois plus de thiosulfate (ñ est le nombre de polysulfuration moyen des ions sulfures). La réaction se fait à chaud (T=70°C) en environ 10 minutes. On complexe ensuite l'excès de sulfite avec du formaldéhyde puis on procède comme pour les dosages précédents. Le soufre de polysulfure est déduit de l'augmentation de la concentration en thiosulfate. Là aussi la concentration du soufre de sulfure doit rester constante ce qui permet de contrôler la précision obtenue dans les dosages successifs.
Pratiquement l'ensemble de ces trois dosages permet donc d'atteindre les quantités suivantes:
- soufre de sulfure : S [H2S] + S [Sn2-] (7)
- soufre de polysulfure : S (n-1) [Sn2-] (8)
- thiosulfate : [S2O32-]
- sulfite : S [SO32-].
Connaissant le pH et la température de l'échantillon, on peut calculer les concentrations en sulfure S [H2S] et en polysulfure S [Sn2-] à partir de (7) et (8).
Pour des concentrations des espèces dissoutes de l'ordre de 10-5 mole.kg-1 on obtient une précision de l'ordre de 1% sur S[H2S], S [Sn2-], S [S2O32-] et S [SO32-]. La précision est de l'ordre de 5% pour des concentrations de l'ordre 10-6 mole.kg-1. En plus d'une précision supérieure, la méthode au chlorure mercurique a aussi l'avantage d'être rapide. En effet avec une burette automatique asservie elle permet de faire l'ensemble des trois dosages en environ 1 heure.
Ces méthodes ont aussi été employées avec succès dans une étude récente des eaux géothermale des Pyrénées Orientales.
c) Comparaison des résultats obtenus sur des eaux par la méthode a l'iode et la méthode aux ions mercuriques.
La comparaison des résultats des dosages des espèces du soufre obtenus par Boulègue au moyen des deux méthodes de dosage sur les eaux sulfurées chaudes des Pyrénées Orientales (voir tableau I) montre que la méthode au chlorure mercurique donne systématiquement des concentrations en thiosulfates significativement inférieures à celles obtenues avec l'iode.
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
||
|
|
|
2,23E-04
|
3,80E-05
|
|
1,84E-04
|
4,00E-06
|
|
||
|
|
|||||||||
|
|
|
1,45E-04
|
1,38E-04
|
|
8,53E-05
|
4,00E-05
|
|
||
|
|
|||||||||
|
|
|
5,00E-05
|
7,00E-06
|
|
4,50E-05
|
6,00E-06
|
|
||
|
|
|||||||||
|
|
|
1,16E-04
|
1,80E-05
|
|
1,38E-04
|
1,00E-05
|
|
||
|
|
|||||||||
Tableau I
Une comparaison plus claire peut apparaître si l'on compare sur des graphiques les résultats obtenus pour la source Beauté entre les deux méthodes.
Comparaison des deux méthodes pour S[H2S] (en mol/l-1 obtenues) :
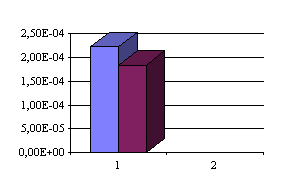
Comparaison des deux méthodes pour [S2O32-] (en mol/l-1 obtenues) :
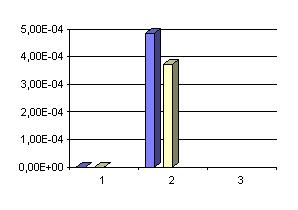
Comparaison des deux méthodes pour 2 S[H2S] +[S2O32-] (en mol/l-1 obtenues) :
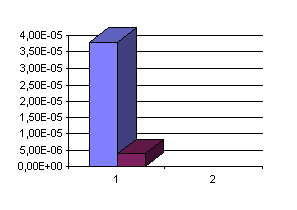
Des résultats semblables sont aussi obtenus si on compare les résultats de Boulègue sur les eaux d'Ax-les-thermes avec ceux d'Auriac, et sur les eaux d'Amélie-les-Bains avec ceux de Baubron et al.(voir tableau II). Comme nous l'avons noté précédemment ces écarts sont attribuables à des pertes d'iode et à la transformation d'iode en iodate au contact des eaux alcalines chaudes des Pyrénées; ce qui conduit à surestimer la concentration en thiosulfate quand on emploie la méthode à l'iode. Dès lors les différences de précision entre les deux méthodes sont clairement mis en évidence lors de ces trois histogrammes: la première méthode ( le dosage avec l'iode est représenté en bleu dans les trois schémas) donne pour tous les dosages effectués des résultats supérieurs à ceux obtenus avec la seconde méthode (avec les ions mercuriques). La méthode employant les ions mercuriques est donc plus précise (dans les eaux chaudes alcalines en tout cas) que celle employant l'iode.
Ces résultats sont confirmés par la spectroscopie d'absorption ultraviolette des eaux. En effet les concentrations en thiosulfate mesurables à 217 nm ne correspondent pas à celles déterminées par la méthode à l'iode mais sont de l'ordre obtenue de celles obtenues avec Hg2+ (la précision de la spectroscopie U.V. à ces faibles concentrations de thiosulfate n'est pas suffisante pour donner une concentration employable du point de vue analytique).
Conclusion
La méthode de dosage des espèces sulfurées, polysulfurées, des thiosulfates et du sulfite par le chlorure mercurique avec une électrode spécifique Ag/Ag2S présente de nombreux avantages par rapport à la méthode à l'iode: précision, fidélité et rapidité. On peut donc proposer d'envisager un remplacement de la méthode à l'iode par la méthode au chlorure mercurique dans les dosages des espèces du soufre dans les eaux sulfurées chaudes. Un des avantages de ce remplacement serait de pouvoir abandonner la notion de "degré sulfhydrométrique" qui n'a pas de fondement physico-chimique du point de vue des relations entre les espèces stables et métastables du soufre dans les eaux sulfurées. Ce serait là un progrès tant du point de vue scientifique que thérapeutique
II) Dosage d'autres éléments des eaux sulfurées:
Les eaux sulfurées ne contiennent pas seulement que des espèces dérivées du soufre. Ainsi de nombreux autres ions sont présents dans la plupart des eaux naturelles. C'est notamment de nombreux cations et de nombreux anions comme le font apparaître les deux compositions des eaux thermales de Cauterets et de Rochefort ( cette dernière nous intéressant particulièrement car nous y effectuerons un dosage dans la suite du devoir).
a) composition de quelques eaux sulfurées.
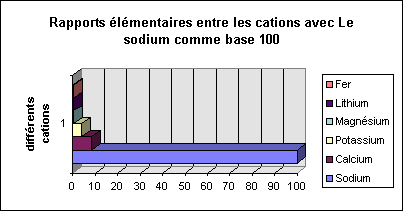
§ Composition de l'eau de Cauterets (source de la Raillère nommée dans les généralités au I) ) dont le pH=9.4, ce qui la classe dans les eaux sulfurées sodiques.
Intéressons nous dans un premier temps à la répartition des cations en g
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ces résultats peuvent apparaître d'une manière plus claire si l'on s'intéresse aux rapports élémentaires entre ces cations.
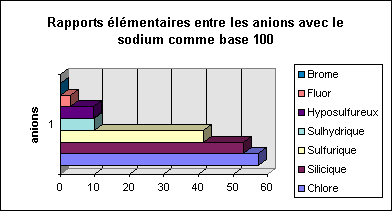
De la même manière intéressons nous aux anions et tout d'abord à la répartition des anions en g
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
En suivant le même principe on peut obtenir une visualisation plus claire de la répartition des anions en s'intéressant aux rapports élémentaires qui existent entre ces différents éléments ( en prenant toujours comme base 100 le cation sodium ce qui permet de simplifier les comparaison entre les anions et les cations).
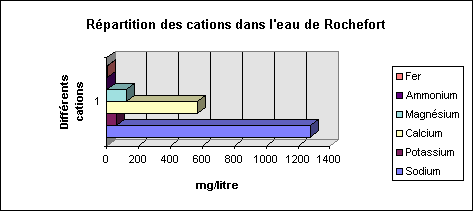
ª Composition de l'eau de Rochefort (Charente Maritime) dont la concentration en fer sera évaluée par la suite. Son pH est de 6.75, ce qui la classe dans les eaux sulfurées calciques neutres.
La répartition des cations se fait selon le tableau suivant :
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ce qui se traduit par le graphique suivant :
De même les anions se répartissent de la manière suivante :
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ce qui se traduit par un graphique

b) Dosage des ions Fe2+ :
PRINCIPE :
Dosage colorimétrique des ions Fe2+ après les avoir complexés avec l'orthophénentroline. La couleur du complexe est rouge, ce qui permet de faire une étude spectrophotométrique.
REACTIFS :
1- Solution d'acide acétique à 1 Mol.l-1.
2- Solution d'orthophénentroline à 275g par litre.
3- Solution d'hydroquinone à 150g par litre.
4- Solution de sulfate de fer à 4.10-4 Mol.l-1.
5- Eau à doser.
MODE OPERATOIRE :
Estimation de la quantité de fer présente dans l'échantillon à doser:
D'après les données qui nous ont été envoyées par la station thermale de Rochefort d'où provient cette eau, la concentration en fer de l'eau dosée 4.2 mg/litre.
En sachant que MFe = 56 g.Mol-1.On en déduit aisément [Fe2+]e = 7.5.10-5 Mol.l-1.
Dès lors on fabrique une gamme de tubes d'étalonnage encadrant cette valeur.
Préparation des échantillons qui serviront à établir la courbe d'étalonnage :
Solution 0: 1 cm3 d'une solution d'acide acétique (1), 2 cm3 de solution d'orthophénentroline (2), 1 cm3 de solution d'hydroquinone (3), 0 cm3 de solution de sulfate de fer (4), le tout versé dans une fiole de 50 cm3 que l'on complète avec de l'eau distillée. Cette solution sera versée dans le Tube 0.
Solution I: 1 cm3 d'une solution d'acide acétique (1), 2 cm3 de solution d'orthophénentroline (2), 1 cm3 de solution d'hydroquinone (3), 2 cm3 de solution de sulfate de fer (4), le tout versé dans une fiole de 50 cm3 que l'on complète avec de l'eau distillée. Cette solution sera versée dans le Tube 1.
Solution II: 1 cm3 d'une solution d'acide acétique (1), 2 cm3 de solution d'orthophénentroline (2), 1 cm3 de solution d'hydroquinone (3), 4 cm3 de solution de sulfate de fer (4), le tout versé dans une fiole de 50 cm3 que l'on complète avec de l'eau distillée Cette solution sera versée dans le Tube 2.
Solution III: 1 cm3 d'une solution d'acide acétique (1), 2 cm3 de solution d'orthophénentroline (2), 1 cm3 de solution d'hydroquinone (3), 6 cm3 de solution de sulfate de fer (4), le tout versé dans une fiole de 50 cm3 que l'on complète avec de l'eau distillée. Cette solution sera versée dans le Tube 3.
Solution IV: 1 cm3 d'une solution d'acide acétique (1), 2 cm3 de solution d'orthophénentroline (2), 1 cm3 de solution d'hydroquinone (3), 8 cm3 de solution de sulfate de fer (4), le tout versé dans une fiole de 50 cm3 que l'on complète avec de l'eau distillée. Cette solution sera versée dans le Tube 4.
Solution V: 1 cm3 d'une solution d'acide acétique (1), 2 cm3 de solution d'orthophénentroline (2), 1 cm3 de solution d'hydroquinone (3), 10 cm3 de solution de sulfate de fer (4), le tout versé dans une fiole de 50 cm3 que l'on complète avec de l'eau distillée. Cette solution sera versée dans le Tube 5.
Préparation de l'échantillon d'eau à doser :
Dans une fiole de 50 cm3, on verse 20 cm3 de l'eau à doser .Puis on verse ensuite 1 cm3 d'une solution d'acide acétique (1), 2 cm3 de solution d'orthophénentroline (2), 1 cm3 de solution d'hydroquinone (3), et on complète avec de l'eau distillée. On versera cette solution dans le Tube 6.
Concentration en fer obtenue dans les tubes d'étalonnage :
Tube 0: [Fe2+] = 0
Tube 1: [Fe2+] = 1.6.10-5 Mol.l-1.
Tube 2: [Fe2+] = 3.2.10-5 Mol.l-1.
Tube 3: [Fe2+] = 4.8.10-5 Mol.l-1.
Tube 4: [Fe2+] = 6.4.10-5 Mol.l-1.
Tube 5: [Fe2+] = 8.0.10-5 Mol.l-1.
Tube 6: on verse l'échantillon à doser.
A ce stade de la manipulation, nous nous rendons compte que le Tube 6 a une coloration qui n'entre pas dans la gamme de couleur des tubes d'étalonnage. En effet le Tube 6 s'avère être bien moins coloré que le Tube 1, qui pourtant contient la quantité la plus faible de fer (hormis le Tube 0) et donc qui devrait être le plus pâle des 7 tubes. On décide donc de poursuivre la manipulation mais en prenant cette fois-ci une solution de sulfate de fer à 5.10-5 Mol.l-1.En suivant le même mode opératoire en ce qui concerne la préparation des tubes d'étalonnage, on arrive aux concentrations suivantes dans les tubes :
Tube 0: [Fe2+] = 0
Tube 1: [Fe2+] = 2.10-6 Mol.l-1.
Tube 2: [Fe2+] = 4.10-6 Mol.l-1.
Tube 3: [Fe2+] = 6.10-6 Mol.l-1.
Tube 4: [Fe2+] = 8.10-6 Mol.l-1.
Tube 5: [Fe2+] = 1.10-5 Mol.l-1.
Pour l'échantillon à doser on procède de la façon suivante
Dans une fiole de 50 cm3, on verse 40 cm3 de l'eau à doser .Puis on verse ensuite 1 cm3 d'une solution d'acide acétique (1), 2 cm3 de solution d'orthophénentroline (2), 1 cm3 de solution d'hydroquinone (3), et on complète avec de l'eau distillée. On versera cette solution dans le Tube 6.
Tube 6: on verse l'échantillon à doser.
Ainsi le Tube 6 à une coloration qui entre dans la gamme de couleur des tubes d'étalonnage.
RESULTATS
Obtention de la courbe d'étalonnage :
On règle le spectrophotomètre à l =510 nm.
Absorbance des tubes :
Tube 0 : 0.
Tube 1 : 0.017.
Tube 2 : 0.043.
Tube 3 : 0.067.
Tube 4 : 0.088.
Tube 5 : 0.114.
Absorbance du Tube 6 : 0.079.
En reportant sur la courbe d'absorbance f ([Fe2+]) on obtient dans le Tube 6 [Fe2+]t=6.95 10-6 Mol.l-1.On en déduit [Fe2+]t = 1.25* [Fe2+]t' =8.68 10-6 Mol.l-1.
Explication de l'échec de la première gamme de tube d'étalonnage:
Nous pouvons expliquer l'échec de la première gamme de tube d'étalonnage, par la durée de stockage de l'eau que nous avons dosé. En effet entre le moment où nous avons reçu l'eau et le moment où le dosage a été effectué s'est écoulé une semaine. Et durant ce laps de temps quasiment tout le fer contenu dans l'eau a du s'oxyder. Conscient de problème nous avons bien essayer d'introduire de nombreux réducteur dans la solution thermale, mais le seul effet notable a été de dissoudre une sorte de suspension rougeâtre que l'on pouvait apercevoir au préalable, mais cependant la concentration en fer obtenue par le dosage n'a pourtant pas augmenter. C'est ce qui explique que [Fe2+]t » 0,115 * [Fe2+]e
c) Dosage Gravimétrique du calcium :
OBJET DE LA NORME
La présente norme a pour objet la description d'une méthode de dosage gravimétrique du calcium dans les eaux.
PRINCIPE
Ce dosage se fait par précipitation de l'oxalate de calcium puis par calcination et enfin par pesée de la chaux.
REACTIFS
1- Acide acétique cristallisable.
2- Acide chlorhydrique (r 20 = 1.19 mg.l-1 ).
3- Ammoniaque (r 20 = 0.925 mg.l-1 ), diluée au dixième.
4- Chlorure d'ammonium : solution à 100 g.l-1.
5- Oxalate d'ammonium : solution à 40 g.l-1.
6- Oxalate d'ammonium : solution à 10 g.l-1.
7- Bleu de bromothymol : solution à 1 g.l-1 dans l'alcool dans 95 % .
MODE OPERATOIRE
Prise d'essai
On prélève 200 à 1000 ml, de façon à ce que l'essai contienne de 10 à 100 mg de calcium.
Préparation de l'échantillon
On neutralise la prise d'essai par l'acide chlorhydrique (2), en présence de bleu de bromothymol (7). Puis on ajoute encore dix gouttes d'acide chlorhydrique (2). On évapore doucement, si nécessaire pour ramener le volume à 200 ml environ et on verse dans un bêcher forme basse de 500 ml.
Dosage
On ajoute 10 ml de chlorure d'ammonium (4) puis lentement l'ammoniaque (3) jusqu'à l'obtention d'une réaction faiblement alcaline ( coloration bleue ) et enfin 10 ml d'acide acétique (1).On fait bouillir 1 minute et on ajoute 50 ml d'oxalate d'ammonium (5).Il faut maintenir l' ébullition 2 minutes puis laisser reposer 2 à 3 heures. On filtre enfin dans un bécher forme haute de 500 ml et on lave avec la solution d'oxalate d'ammonium chaude (6).On introduit le filtre humide dans un creuset de platine taré. On sèche par chauffage modéré, on incinère avec précaution le filtre et on calcine à 1000°C jusqu'à masse constante. On pèse le résidu.
Expression des résultats
Soient :
V le volume en millilitres de la prise d'essai.
M la masse en milligrammes de la chaux.
La teneur en calcium exprimée en milliéquivalent (mé) au litre est donnée par l'expression :
![]() soit
soit ![]()
Il est rappelé que : 1 mé au litre = 20.04 mg au litre d'ions Ca2+.
1 mg au litre = 0.05 mé d'ion Ca2+.